


The Impact of the Williams Syndrome Mutations on Neural James S. McDonnell Foundation Collaborative Activity Award: |
Project 2: Are certain genes deleted in Williams syndrome specifically related to sociability? Search strategy and molecular characterization of Williams individuals with different size deletions (Korenberg and Bellugi).
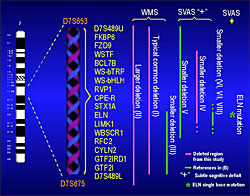 Bellugi and Korenberg have established an extensive network of potential Williams subjects by working with the Williams Syndrome Association, hosting conferences for parents, writing for their newsletters, sharing results, and helping to turn research into practice for families of individuals with Williams. Through this network of established contacts, we will be able to locate and induct into our ongoing studies not only Williams individuals with classical deletions, but also newly discovered rare individuals with smaller deletions, some of whom are indicated in the figure. We already have located a dozen or so Williams individuals who have different or smaller size deletions, and preliminary molecular analysis indicates that some are missing all but two or three specific genes at the end of the deletion (Korenberg, et al, 1999, 2000; 2001). These individuals with deletions that affect a contiguous region including three genes, CYLN2, GTF2IRD1 and GTF2I, appear to differ from full deletion Williams in their social behavior; based on preliminary observations they do not appear to exhibit the hypersociability of typical Williams. If this is borne out in these and additional subjects, it would suggest that the deletion of only 1-3 genes produces the profound change in social behavior that is so typical of classical Williams syndrome.
Bellugi and Korenberg have established an extensive network of potential Williams subjects by working with the Williams Syndrome Association, hosting conferences for parents, writing for their newsletters, sharing results, and helping to turn research into practice for families of individuals with Williams. Through this network of established contacts, we will be able to locate and induct into our ongoing studies not only Williams individuals with classical deletions, but also newly discovered rare individuals with smaller deletions, some of whom are indicated in the figure. We already have located a dozen or so Williams individuals who have different or smaller size deletions, and preliminary molecular analysis indicates that some are missing all but two or three specific genes at the end of the deletion (Korenberg, et al, 1999, 2000; 2001). These individuals with deletions that affect a contiguous region including three genes, CYLN2, GTF2IRD1 and GTF2I, appear to differ from full deletion Williams in their social behavior; based on preliminary observations they do not appear to exhibit the hypersociability of typical Williams. If this is borne out in these and additional subjects, it would suggest that the deletion of only 1-3 genes produces the profound change in social behavior that is so typical of classical Williams syndrome.
The genes GTF2I and GTF2IRD1 are expressed throughout the body in adults but, beyond a knowledge of expression related to immune activation, little information is available on their overall developmental expression or on the expression of transcriptional or post translational variants in either the fetal or adult brain. Preliminary studies indicate that, based on its unique physical and functional interactions at both the Inr element and upstream regulatory sites, GTF2I is postulated to be a novel transcriptional cofactor that integrates signals from the regulatory components to the basal machinery. It was recently shown that GTF2I is phosphorylated at both serine and tyrosine residues and that tyrosine phosphorylation of GTF2I is critical for its transcriptional properties. Equally interesting is the observation that a variety of extracellular signals mediated through cell surface receptors, including growth factor receptors, lead to enhanced tyrosine phosphorylation and increased transcriptional activity of GTF2I (Desiderio & Wang, 1999). GTF2IRD1 encodes a protein of 944 amino acids that contains a region of high similarity to a unique motif with helix-loop-helix forming potential, that also occurs within the transcription factor GTF2I. Analogous to GTFII-I, the product of GTF2IRD1 may have the ability to interact with other HLH-proteins and function as a transcription factor or as a negative transcriptional regulator (Franke, Peoples, & Francke, 1999). Moreover, these related transcriptional regulators are of particular interest in that in some parts of the brain, they are thought to be co-regulators. Data are sorely lacking in all developmental systems and these genes would be particularly useful for establishing and manipulating an animal model capable of higher cognitive processes such as the rhesus monkey. The gene for CYLN2 (alias CLIP-115), located just centromeric to the GTF2I related genes, is also of significant interest. Cytoplasmic linker proteins (CLIPs) have been proposed to mediate the interaction between specific membranous organelles and microtubules. Recently, this gene was characterized as a novel member of this family, called CLIP-115. This protein is most abundantly expressed in the brain and was found to associate both with microtubules and with the dendritic lamellar body (Hoogenraad et al., 1998).
To define the genes deleted and their distances from the deletion breakpoint in Williams subjects with putatively smaller deletions, chromosome preparations and DNA isolated from cell lines will be analyzed using fluorescence in situ hybridization of human BACs carrying specific genes from the Williams region (Korenberg et al., 2001) and quantitative Southern blot dosage analyses. Because some small deletions may be complex and may skip or rearrange some genes, it is important to confirm exact breakpoints and sample the entire region that is deleted in typical Williams syndrome. For subjects with unique, small deletions, somatic cell hybrids containing the deleted chromosome 7 will be generated to facilitate DNA sequence analysis in the absence of the normal chromosome 7. These analyses will result in the accurate definition of breakpoints to within 10 kb, providing a precise definition of the genes and control regions deleted. They will also identify those genes neighboring the breakpoint whose expression may be altered and will be further candidates related to the hypersociality observed in Williams syndrome.
In summary, the major specific aim of Project 2 will be to contrast the molecular genetics of Williams individuals with typical versus smaller deletions, in order to elucidate the gene or genes that may be associated with the prominent hypersocial behavior typical of the full deletion Williams subject. This project includes both molecular genetic analysis of Williams individuals with different size deletions and integration with behavioral characterization above.
| Introduction | Project 1 | Project 2 | Project 4 | Project 5 | Project 6 | Conclusion |